
ERLEADA(apalutamide)使用说明书
处方资料重点
这些重点不包括安全和有效使用ERLEADA需所有资料。请参阅ERLEADA完整处方资料。
ERLEADATM(apalutamide)片,为口服使用
美国初次批准 – 2018
适应证和用途
ERLEADA是一种雄激素受体抑制剂适用为有非-转移去势-抗性前列腺癌患者的治疗。(1)
剂量和给药方法
ERLEADA 240 mg(四60 mg片)每天一次口服给予。整吞片。ERLEADA可被有或无食物服用。(2.1)
患者还应接受一种促性腺激素释放激素(GnRH)类似物同时地或应曽有双侧睾丸切除。(2. 1)
剂型和规格
片: 60 mg(3)
禁忌证
妊娠。(4,8.1)
警告和注意事项
●接受ERLEADA患者分别发生跌倒和骨折16%和12%。评价患者对骨折和跌倒风险,和根据已确定指导原则用骨靶向药剂治疗患者。(5.1)
●癫痫发生在0.2%的接受ERLEADA患者。治疗期间发生癫痫的患者永久地终止ERLEADA。(5.2)。
不良反应
最常见不良反应(≥10%)为疲乏,高血压,皮疹,腹泻,恶心,体重减轻,关节痛,跌倒,热潮红,食欲减低,骨折,和周围水肿。(6.1)
报告怀疑不良反应,联系JanssenProducts,LP电话1-800-526-7736(1-800-JANSSEN或FDA电话1-800-FDA-1088或www.fda.gov/medwatch.
药物相互作用
●与药物是CYP3A4,CYP2C19,CYP2C9,UGT,P-gp,BCRP,或OATP1B1的敏感底物同时使用可能导致这些药物丧失活性。(7.2)
在特殊人群中使用
●生殖潜能的女性和男性: 建议男性有生殖潜能女性伴侣的男性使用有效避孕。(8.3)
完整处方资料
1 适应证和用途
ERLEADA是适用为有非-转移,去势-抗性前列腺癌(NM-CRPC)患者的治疗。
2 剂量和给药方法
2.1 推荐剂量
ERLEADA的推荐剂量为240 mg(四60 mg片)每天一次口服给予。整吞片。ERLEADA可被有或无食物服用。
患者还应接受一个促性腺激素释放激素(GnRH)类似物同时地或应曽有双侧睾丸切除。
2.2 剂量修饰
如一位患者经受一个大于或等于级别3毒性或一个不能耐受副作用,不给药直至症状改善至低于级别1或原始级别,然后在相同剂量或一个减低剂量(180mg或120 mg),如断定。
3 剂型和规格
片(60 mg): 略微黄至灰绿色椭圆薄膜-包衣片,在一侧凹陷有“AR 60”。
4 禁忌证
妊娠。
ERLEADA可能致胎儿危害和潜在丢失妊娠[见在特殊人群中使用(8.1)]。
5 警告和注意事项
5.1 跌倒和骨折
接受ERLEADA患者中发生跌倒和骨折。评价患者对骨折和跌倒风险。根据已确定的治疗指导原则监视和处置患者处于风险骨折和考虑使用靶向骨药剂。
在一项随机化研究(SPARTAN),在16%用ERLEADA治疗患者发生跌倒与之比较用安慰剂治疗9%患者。跌倒是不伴随意识丧失或癫痫。用ERLEADA治疗患者发生骨折12%和用安慰剂治疗患者发生7%。用ERLEADA治疗患者发生3%的级别3-4骨折和用安慰剂治疗患者发生1%。对用ERLEADA治疗患者骨折发病的中位时间为314天(范围:20至953天)。在SPARTAN研究未进行常规骨密度评估和用靶向骨剂骨质疏松的治疗。
5.2 癫痫
接受ERLEADA患者中发生癫痫。期间发生癫痫的患者永久终止ERLEADA。不知道用ERLEADA抗-癫痫药物是否将预防癫痫。忠告患者当接受ERLEADA发生一个癫痫的风险和从事任何活动期突然丧失意识可能致危害于他们自身或他人。
在一项随机化研究(SPARTAN),两例用ERLEADA治疗的患者(0.2%)经受一个癫痫。
癫痫发生于从ERLEADA的开始后354至475天。用安慰剂治疗患者没有发生癫痫。
排除有癫痫病史,对癫痫易发生因子,或接受药物已知减低癫痫阈值或诱发癫痫的患者。
对于在再给予ERLEADA 予至经受一次癫痫患者没有临床经验。
6 不良反应
说明书的其他节中更详细讨论以下:
● 跌倒和骨折[见警告和注意事项(5.1)]
● 癫痫[见警告和注意事项(5.2)]
6.1 临床试验经验
因为临床试验是在广泛不同条件下进行,在一种药物临床试验观察到的不良反应率不能直接地与另一种药物临床试验率比较和可能不反映在实践中观察到的发生率。
SPARTAN,一项随机化(2:1),双盲,安慰剂-对照,多-中心临床研究,纳入有非-转移,去势-抗性前列腺癌(NM-CRPC)患者。在这项研究中,患者接受或ERLEADA在一个剂量240mg每天或一种安慰剂。在SPARTAN研究所有患者接受一种同时促性腺激素释放激素[促性腺激素释放激素(GnRH)]类似物或有一个双侧睾丸切除。
在接受ERLEADA患者暴露的中位时间为16.9月(范围: 0.1至42月)和接受安慰剂患者中为11.2 月(范围:0.1至37月)。总体而言,用ERLEADA治疗8例患者(1%)死于不良反应。对死亡理由为感染(n=4),心肌梗死(n=3),和脑出血(n=1)。一例患者(0.3%)treated 用安慰剂治疗死于一个心肺停止不良反应(n=1)。在11%ERLEADA患者被终止由于不良反应,最常见来自皮疹(3%)。不良反应导致给药中断或ERLEADA的减低发生在33%患者;最常见(>1%)为皮疹,腹泻,疲乏,恶心,呕吐,高血压,和血尿。严重的不良反应发生在25%的ERLEADA-治疗患者而接受安慰剂患者的23%。在ERLEADA臂最常见严重的不良反应(>2%)为骨折(3%)和在安慰剂臂为尿潴留(4%)。表1 显示在SPARTAN不良反应发生在≥10%用ERLEADA臂发生频数与安慰剂比较绝对增高2%。表2显示实验室异常发生在≥15%的患者,和在ERLEADA臂比安慰剂更频(>5%)。
附加临床意义不良反应发生在 2%或更多用ERLEADA治疗患者包括甲状腺功能减退(8.1%相比 2%用安慰剂),瘙痒(6.2%相比用安慰剂2%),缺血性心脏病(3.7% 相比 用安慰剂2%),和心衰(2.2%相比用安慰剂1%)
皮疹
在SPARTAN中,皮疹伴随ERLEADA被最常见被描述为斑点或班-丘疹。用ERLEADA治疗患者皮疹不良反应被报道为24% of相比用安慰剂治疗患者6%。用ERLEADA治疗被报道(5%)级别3皮疹(被定义为覆盖>30%体表面积[BSA])相比安慰剂(0.3%)。皮疹的开始发生ERLEADA治疗从皮疹开始一个中位82天。皮疹解决在81%的患者中位60天(范围:2至709天)。为治疗皮疹,四例(4%)用ERLEADA治疗患者接受全身皮质激素。在大约地半数患者皮疹再发生被用ERLEADA再次挑战。
甲状腺功能减退
用ERLEADA治疗患者甲状腺功能减退被报道为8%而用安慰剂治疗患者2%。根据每4月甲状腺-刺激激素(TSH)评估。在25%用ERLEADA治疗患者发生升高的TSH和用安慰剂治疗患者为7%。中位开始时间为天113。没有级别3或4不良反应。用ERLEADA治疗患者7%开始甲状腺替代治疗。甲状腺替代治疗,当临床有适应证时,应被开始或剂量-调整[见药物相互作用(7.2)]。
7 药物相互作用
7.1其他药物对ERLEADA的影响
强CYP2C8或CYP3A4抑制剂的共同给药一种强CYP2C8或CYP3A4抑制剂被预期增加活性部分的稳态暴露(非结合apalutamide加效力-调整非结合的N-desmethyl-apalutamide之和)。
无需初始剂量调整但是,根据耐受性减低ERLEADA剂量[见剂量和给药方法(2.2)]。CYP2C8或CYP3A4的轻度或中度抑制剂是不被预期影响apalutamide的暴露。
7.2 ERLEADA对其他药物的影响
CYP3A4,CYP2C9,CYP2C19和UGT底物在人中,ERLEADA是CYP3A4和CYP2C19一种强诱导剂,,和CYP2C9的一种弱诱导剂。同时使用 ofERLEADA与药物主要地被CYP3A4,CYP2C19,或CYP2C9代谢药物可能导致这些药物的较低暴露。如药物被继续,当对活性丧失可能性或评价建议取代这些药物。ERLEADA与药物的共同给药是UDP-葡萄醛酸基移换脢(UGT)的底物可能导致减低暴露。谨慎使用如UGT的底物必须与ERLEADA共同给药和评价对活性的丧失[见临床药理学(12.3)]。
P-gp,BCRP或OATP1B1底物Apalutamide被显示是P-糖蛋白(P-gp),乳癌抗性蛋白(BCRP),和有机阴离子转运多肽1B1(OATP1B1)临床上一种弱诱导剂。在稳态时,apalutamide减低对非索非那定[fexofenadine](一种P-gp底物)血浆暴露和瑞舒伐他汀[rosuvastatin](一种BCRP/OATP1B1底物)。ERLEADA与药物是P-gp,BCRP,或OATP1B1底物的同时使用可能导致这些药物的较低暴露。谨慎使用如P-gp,BCRP或OATP1B1的底物必须与ERLEADA共同给药和评价活性丧失如药物是继续使用[见临床药理学(12.3)]。
8 在特殊人群中使用
8.1 妊娠
风险总结
ERLEADA被禁忌在妊娠妇女使用因为药物可能致胎儿危害和妊娠潜在丢失。ERLEADA是不适用在女性中使用,所以没有用apalutamide进行动物胚胎-胎儿发育毒理学研究。没有在妊娠妇女中使用ERLEADA的人数据。根据它的作用机制,当妊娠期间给予ERLEADA可能致胎儿危害。
8.2 哺乳
风险总结
ERLEADA是不适用为女性使用。对apalutamide或它的代谢物在人乳汁中存在,对哺乳喂养儿童的影响,或对乳汁产生的影响没有数据。
8.3 生殖潜能的女性和男性
避孕
男性
根据作用机制和在动物生殖研究中发现,建议有生殖潜能女性伴侣男性患者治疗期间和ERLEADA末次剂量后共3个月使用有效避孕。.[见在特殊人群中使用(8.1)].
不孕不育
男性
根据动物研究,ERLEADA可能损害在生殖潜能男性中生育力[见肺临床毒理学(13.1)]。.
8.4 儿童使用
尚未确定在儿童患者中ERLEADA的安全性和有效性。
8.5 老年人使用
在SPARTAN中接受ERLEADA的803例患者,87%的患者为65岁和以上和49%为75岁和以上。级别3-4不良反应发生在46%(323/ 697)的患者65岁或以上和在51%(197/391)的患者75岁或以用ERLEADA治疗与用安慰剂治疗比较,35%(124/355)患者65岁或以上和37%(70/187)的患者75岁或以。这些患者和较年轻患者间未观察到有效性总体差别。
10 药物过量
对apalutamide药物过量没有已知的特异性抗毒剂。在药物过量的事件中,停止ERLEADA,采用一般性支持性措施直至临床毒性已被消除或解决。
11 一般描述
Apalutamide,ERLEADA的活性成分,是一种雄激素受体抑制剂。化学名为(4-[7-(6-Cyano-5-trifluoromethylpyridin-3-yl)-8- oxo-6- thioxo-5,7-diazaspiro[3.4]oct-5- yl]-2-fluoro-N-methylbenzamide)。
Apalutamide是白色至略微黄色粉。Apalutamide是实际上不溶于水性介质跨越一个广泛的pH值。分子量为477.44和分子式为C21H15F4N5O2S。结构式为:
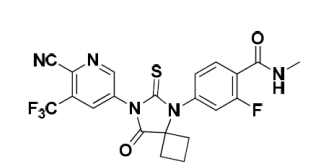
ERLEADA(apalutamide)被供应为膜-包衣片为口服给药含60mg的apalutamide。核心片的无活性成分为无水胶体二氧化硅,交联羧甲基纤维素钠,羟丙基甲基纤维素-醋酸琥珀,硬脂酸镁,微晶纤维素,和硅化微晶纤维素。该片用一种商品可得到的膜包衣组成完成,由以下赋形剂组成:氧化铁黑,氧化铁黄,聚乙二醇,聚乙烯醇,滑石,和二氧化钛。
12 临床药理学
12.1 作用机制
Apalutamide是一种雄激素受体(AR)抑制剂直接地结合至AR的配体-结合结构域。Apalutamide抑制AR核转位,抑制DNA结合,和妨碍AR -介导的转录。一个主要代谢物,N-desmethylapalutamide,是一个效力较弱AR抑制剂,和表现出apalutamide活性的三分之一在一项体外转录受体分析。Apalutamide给药致减低的肿瘤细胞增殖和增加凋亡导致减低肿瘤容积在前列腺小鼠移植物模型。
12.2 药效动力学
心脏电生理学
在一项开放,无对照,多中心,单-臂专门QT研究在45例有CRPC患者评估apalutamide 240mg每天1次对QTc间期的影响。从基线最大均数QTcF变化为12.4 ms(2-侧90%上CI: 16.0ms)。在QTcF对apalutamide和它的活性代谢物一项暴露-QT分析提示一个浓度-依赖增加。.
12.3 药代动力学
Apalutamide药代动力学参数被展示为均数[标准差(SD)]除非否则说明。重复每天一次给药30至480mg后(0.125至2倍推荐剂量)后一palutamideCmax和浓度曲线下面积(AUC)正比例地增加。推荐剂量的给药后4周后,apalutamide达到稳态和均数虚机比值为大约5-倍。在稳态时Apalutamide Cmax为6.0 µg/mL(1.7)和AUC为100µg·h/mL(32)。Apalutamide血浆浓度每天波动为低,有均数峰-至-谷比值1.63。用重复给药观察到一个表观清除率(CL/F)增加,可能由于apalutamide的自身诱导代谢。
在推荐剂量时自身-诱导效应可能达到它的最大值。因为apalutamide跨越剂量范围30至480mg是剂量-正比例。在稳态时在推荐剂量后,主要活性代谢物N-desmethyl apalutamide Cmax为5.9µg/mL(1.0)和AUC为124 µg·h/mL(23)。在稳态时,N-desmethylapalutamide特征被一个平坦浓度-时间图形描述有一个均数峰-至-谷比值为1.27。对N-desmethylapalutamide 重复给药后均数AUC代谢物/母药比值为1.3。根据全身暴露,相对效力,和药代动力学性质,N-desmethylapalutamide 可能对apalutamide临床活性有贡献。
吸收
均数绝对口服生物利用度为大约100%。达到血浆峰浓度(tmax)中位时间为2小时(范围: 1至5 小时)。
食物的影响
Aapalutamide的给药对健康受试者空腹条件下和与一个高-脂肪餐(大约500至600脂肪卡路里,250碳水化合物卡路里,和150蛋白质卡路里)导致无临床相关变化在Cmax和AUC。有食物到达tmax中位时间被延迟大约2小时。
分布
Apalutamide在稳态时均数表观分布容积为大约276 L。
结合至血浆蛋白Apalutamide为96%和N-desmethyl apalutamide为95%与无浓度依赖性。
消除
单次给药后apalutamide的CL/F为1.3 L/h和在每天一次给药后在稳态时增加至 2.0L/h可能是由于CYP3A4自身-诱导作用。在稳态时在患者中对apalutamide均数有效半衰期为大约3天。
代谢
代谢是apalutamide的消除主要途径。Apalutamide是主要地被CYP2C8和CYP3A4代谢形成活性代谢物,N-desmethylapalutamide。在单次给药后,apalutamide的代谢中CYP2C8和CYP3A4的贡献被估计分别是58%和13%但在稳态时变化至40%和37%。
在单次口服放射性标记的apalutamide 240 mg后, Apalutamide代表总AUC的45%和N-desmethylapalutamide代表44%。排泄直至单次口服给予放射标记apalutamide后70天,在尿中回收65%的剂量(1.2%的剂量为未变化的apalutamide和2.7%为N-desmethylapalutamide)和24%在粪中被回收(1.5%的剂量为未变化的apalutamide和2%为N-desmethylapalutamide)。
特殊人群
根据年龄(18-94岁),种族(黑种人,肺-日本亚裔,日本人),轻度至中度(eGFR 30-89mL/min/1.73m2,膳食在肾病[MDRD]方程的修饰估算)肾受损,或轻度(Child-Pugh A)至中度(Child-Pugh B)肝受损未观察到apalutamide或N-desmethylapalutamide的药代动力学临床意义差别。
不知道严重肾受损或肾病终末期(eGFR ≤29mL/min/1.73m2,MDRD)或严重肝受损(Child-PughC)对apalutamide 药代动力学的影响。
药物相互作用
其他药物对ERLEADA的影响
强CYP2C8抑制剂
ERLEADA作为一个240mg单次给药与吉非罗齐[gemfibrozil](一种强CYP2C8抑制剂)的共同给药后ApalutamideCmax减低21%而AUC增高 68%。吉非罗齐被期望增加稳态apalutamide Cmax 32%和AUC44%。对活性部分(非结合的apalutamide加效力-调整的非结合的N-desmethylapalutamide之和),预期的稳态Cmax增加19%和AUC 23%。
强CYP3A4抑制剂
Apalutamide Cmax减低22%而对ERLEADA作为一个240mg单次给药与伊曲康唑[itraconazole](一种强CYP3A4抑制剂)的共同给药后AUC相似。酮康唑[Ketoconazole](一种强CYP3A4抑制剂)被预期增加单次-给药apalutamideAUC 24%但对Cmax没有影响。酮康唑被期前增加稳态apalutamide Cmax至38%和AUC51%。对活性部分,期望的稳态Cmax增加23%和AUC 28%。
CYP3A4/CYP2C8诱导剂
利福平[Rifampin](一种强CYP3A4和中度CYP2C8诱导剂)被预期减低稳态apalutamide Cmax 25%和AUC 34%。对活性部分期望的稳态Cmax减低15%和AUC 19%。
酸降低剂
Apalutamide在相关生理学pH条件下是不离子化[ionizable],所以酸降低剂(如,质子泵抑制剂,H2-受体拮抗剂,抗酸剂)不期望影响apalutamide溶解度和生物利用度。在体外药物影响转运蛋白,apalutamide和N-desmethylapalutamide 对 P-gp是底物但BCRP,OATP1B1,和OATP1B3不是。因为apalutamide口服后被完全吸收,P-gp不限制apalutamide吸收和所以,P-gp的抑制作用或诱导作用期望不影响apalutamide的生物利用度。
ERLEADA对其他药物的影响
CYP底物
在体外研究中显示apalutamide和N-desmethylapalutamide为中度至强CYP3A4和CYP2B6诱导剂,是CYP2B6和CYP2C8的中度抑制剂,和CYP2C9,CYP2C19,和CYP3A4的弱抑制剂。在治疗相关浓度时Apalutamide和N-desmethylapalutamide不影响CYP1A2和CYP2D6。ERLEADA与单次口服剂量的敏感CYP底物的共同给药导致一个92%减低在咪达唑仑[midazolam]的AUC(一种CYP3A4底物),奥美拉唑[omeprazole]的AUC的85%减低(一种CYP2C19底物),和S-华法林[warfarin]AUC的46%减低(一种CYP2C9底物)。ERLEADA不致对一种CYP2C8底物暴露中临床上显著变化。
P-gp,BCRP和OATP1B1底物
ERLEADA与单次口服剂量的转运蛋白底物的共同给药导致非索非那定(一种P-gp底物)AUC的一个30%减低和瑞舒伐他汀(一种BCRP/OATP1B1底物)的AUC中41%减低但对Cmax没有影响。
UGT底物
Apalutamide可能诱导UGT。ERLEADA与药物是UGT 的底物的共同给药可能导致OCT2,OAT1,OAT3和MATEs底物这些药物对这些药物较低暴露。在体外,apalutamide和N-desmethylapalutamide抑制有机阳离子转运蛋白2(OCT2),有机阴离子转运蛋白3(OAT3)和多药和毒素挤出[extrusions](MATEs),和不抑制有机阴离子转运蛋白1。Apalutamide对一个OAT3底物暴露预期不致临床意义变化。
13 非临床毒理学
13.1 癌发生,突变发生,生育力受损
未曽进行长期动物研究评价apalutamide的致癌性潜能。Apalutamide不诱发突变在细菌回复突变(Ames)试验和没有遗传毒性在或体外染色体畸变分析或体内大鼠骨髓微核试验或体内大鼠Comet试验。在雄性大鼠重复-给药毒性研究(直至26周)和犬(直至39周),前列腺和贮精囊的萎缩,无精子/精子低下,观察到精子细管退行性变性和/或在生殖系统中间质细胞的增生或肥大在≥25 mg/kg/day在大鼠(根据 AUC人暴露的1.4 倍)和≥2.5 mg/kg/day在犬(根据AUC人暴露0.9倍)。在雄性大鼠中一项生育力研究中,精子浓度和活动性中一个减低,增加异常精子形态学,较低交配和生育力率(与未处理雌性配对)与第二性腺和附睾的重量减低在一起≥25mg/kg/day给药后(根据 AUC人暴露0.8倍)4周观察到。一个减低数量的或胎儿由于增加植入丢失前后被观察到150mg/kg/day给药4周后(根据 AUC人暴露5.7倍)。从末次apalutamide给药后8周后对雄性大鼠效应是可逆性。
14 临床研究
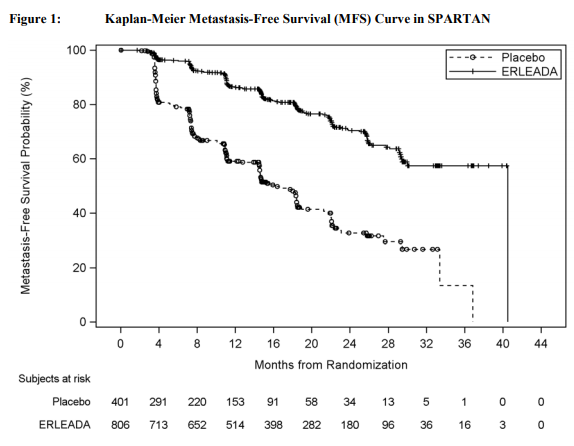
SPARTAN(NCT01946204)是一项多中心,双盲,随机化(2:1),安慰剂-对照临床试验其中1207例有NM-CRPC患者被随机化(2:1)接受或ERLEADA口服在一个剂量240mg每天1次(N = 806)或安慰剂每天1次(N =401)。在SPARTAN试验所有患者接受一个同时和被盲态独立中央审评(BICR)非转移疾病阿确证。PSA结果被盲态和不被用于对治疗终止.患者随机化至或臂终止治疗对放射影像疾病进展BICR确证,仅局部区域进展,新治疗的开始,不能接受的毒性,或撤出.
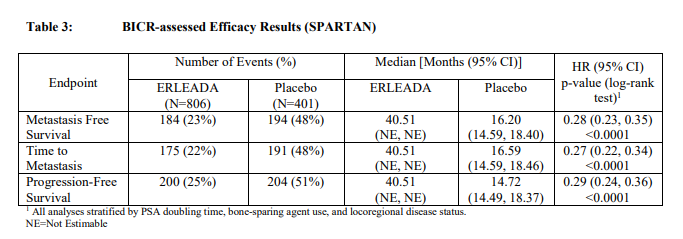
治疗臂间以下患者人口统计和基线疾病特征被平衡。中位年龄为74岁(范围 48-97)和26%的患者为80岁或以上。种族分布为66%高加索人,12%亚裔,和6%黑种人。在两治疗臂77%的患者都有前列腺的以前手术或放射治疗。患者的多数有一个Gleason评分7或更高(78%)。15%的患者有<2cm骨盆淋巴结在研究纳入时。73%的患者接受以前用一种抗雄激素治疗;69%的患者接受比卡鲁胺[bicalutamide]和10%的患者接受氟他胺[flutamide]。在研究纳入时,所有患者有一个美国东部肿瘤合作组肿瘤性能状态[EasternCooperative Oncology Group Performance Status(ECOGPS)]评分为0或1。在终止研究治疗患者中(对安慰剂N = 279和对ERLEADA N =314),用ERLEADA治疗患者(56%)和一个更大比例(80%)的用安慰剂治疗的患者接受随后治疗。总体而言,在2%的患者仅发生局部区域-进展。研究的主要疗效结局测量为无转移生存(MFS),被定义为从随机化至被BICR-确证远部转移的首次证据的时间,被定义为新的骨或软组织病变或增大淋巴结髂总动脉杈[iliacbifurcation]以上,或由于任何原因的死亡,看哪个先发生[whichever occurredfirst]。附加疗效终点为至转移时间(TTM),无进展生存(PFS)其中也包括局部区域进展,至症状进展时间,和总体生存(OS)。在MFS一个统计学上显著改善被证实在患者随机化至接受ERLEADAcompared 与患者随机化至接受安慰剂比较。跨越患者亚组包括PSADT(≤ 6 个月或>6个月),一个以前骨-sparing剂的使用(是或否),和局部区域疾病(N0或N1)被观察到一致结果。主要疗效结局被在TTM,PFS,和至症状进展时间统计学上显著改善支持。在最后MFS分析时总体生存(OS)数据是不成熟(24%的需要事件数)。在图1和表3中总结来自SPARTAN试验MFS,TTM,和PFS的疗效结果。
16 如何供应/贮存和处置
ERLEADA(apalutamide) 60 mg膜包衣片为略微黄至灰绿色椭圆形片在一侧凹陷有“AR 60”。 ERLEADA 60mg片是可得到在120片瓶。每瓶含硅胶干燥剂。
NDC 号59676 600
12 贮存和处置
贮存在20°C至25°C(68°F至77°F); 外出允许至15°C至30°C(59°F至86°F)[见USP控制室温]。
贮存在原始包装,。不要遗弃干燥剂。避光和潮湿保护。
17 患者咨询资料
建议患者阅读FDA-批准的患者说明书(患者信息)。
跌倒和骨折
●告知患者ERLEADA 是伴随一个跌倒和骨折增加发生率[见警告和注意事项(5.1)]。
癫痫
告知患者ERLEADA曽伴随一个癫痫的增加风险。讨论可能易于癫痫条件和可能降低癫痫阈值药物。忠告患者从事在任何活动其中突然丧失意识可能致严重的危害至他们自身或其他人。告知患者如他们经受一个癫痫立即联系他们的卫生保健提供者[见警告和注意事项(5.2)].
皮疹
●告知患者ERLEADA是伴随皮疹和告知他们的卫生保健提供者如他们发生一个皮疹[见不良反应(6.1)]。
剂量和给药方法
● 告知患者接受同时促性腺激素释放激素(GnRH)类似物治疗用ERLEADA治疗的疗程期间需要维持这个治疗。
●指导患者采用他们的剂量在每天相同时间(每天1次)。ERLEADA可被有或无食物服用。 每片应被整吞。
●告知患者在缺失ERLEADA每天剂量的事件中,他们应尽可能马上服用他们的正常剂量在相同天与至在下一天的正常时间表。患者不要服用额外片组成丢失剂量[见剂量和给药方法(2.1)]。
胚胎-胎儿毒性
●告知患者ERLEADA可能危害至发育中胎儿。忠告有生殖潜能女性伴侣治疗期间有性活动患者和ERLEADA的末次剂量后共3个月使用有效避孕。建议男性患者如与一位妊娠妇女有性活动使用一个阴茎套[见在特殊人群中使用(8.1,8.3)]。
不孕不育
●忠告男性患者ERLEADA可能损害生育力和治疗期间和ERLEADA的末次剂量后共3个月不供精子[见在特殊人群中使用(8.3)]。